
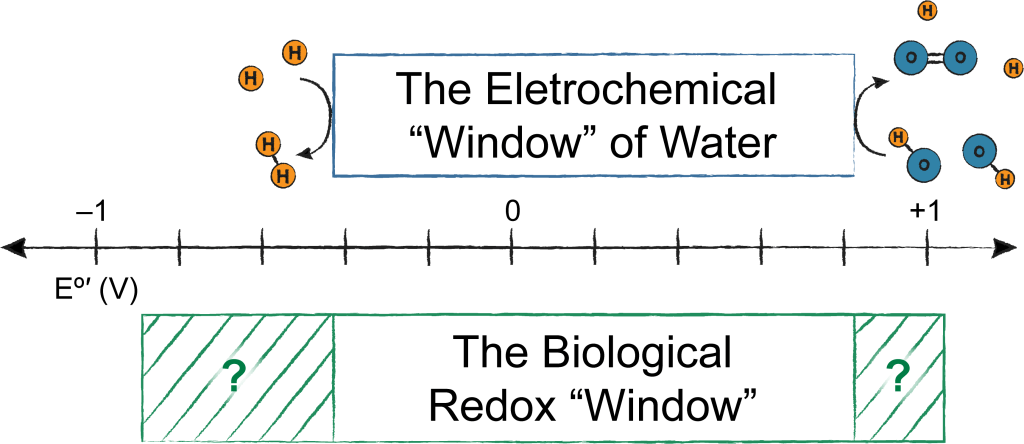
Life happens in water, and while water is a pretty amazing solvent, it can break down at extreme electrochemical potentials, making catalyzing reactions outside this electrochemical “window” challenging and non-selective. It appears biology never got the message; enzymes found in all organisms on the planet carry out reduction/oxidation (redox) reactions that stray outside this electrochemical window and are therefore non-equilibrium processes. We are generally interested in how this is achieved through synergistic properties of enzyme structure, cofactors, and protein dynamics. All redox reactions in this “forbidden territory” require judicious control of electron and proton dynamics, and thus inspire chemical innovations outside the enzyme active site. Some specific projects of interest are highlighted below.
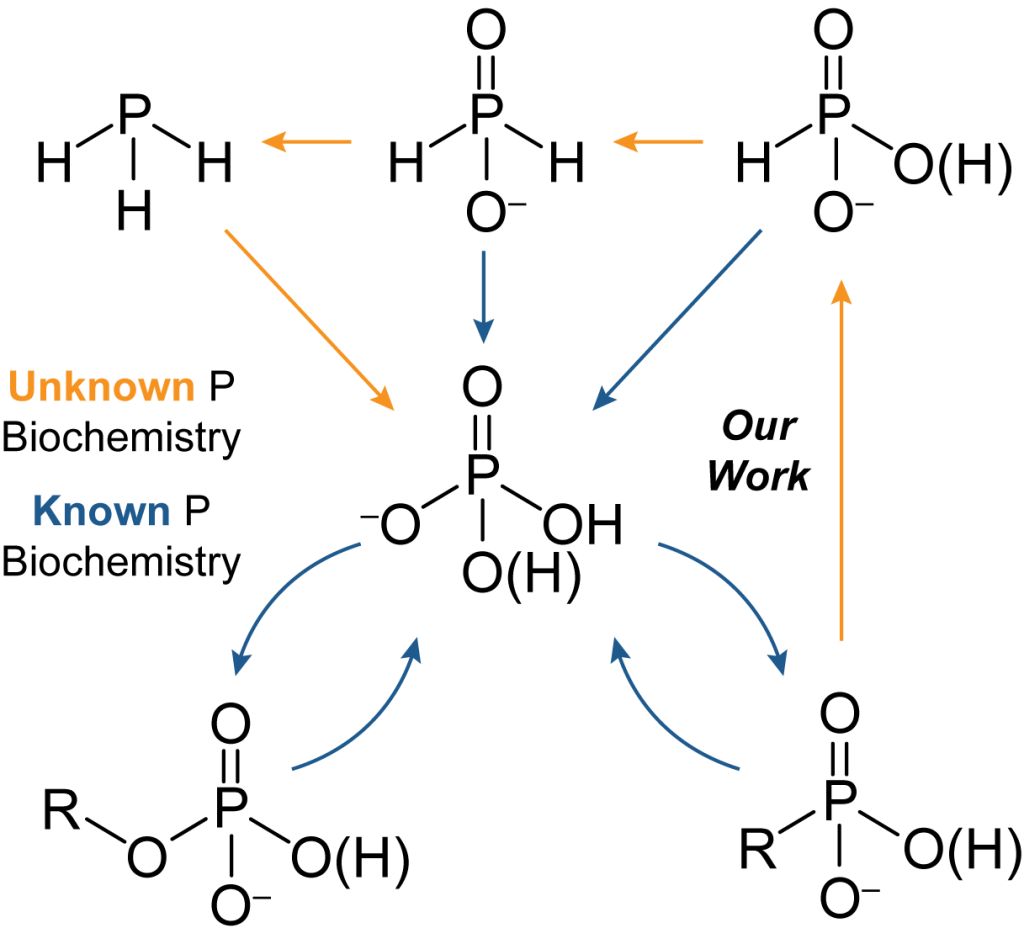
Phosphorus in biological systems is generally assumed to exist exclusively as the phosphate ion, or phosphoesters thereof, and functions solely in acid-base chemistry. Growing evidence suggests that phosphorus redox cycling may be a globally significant process linked to the carbon cycle. We are interested in microbial reduction of phosphate from an energetic, mechanistic, and regulatory perspective. Discoveries in this area have direct implications in the global biogeochemical phosphorus cycle, and may present novel green synthetic routes to reduced phosphorus compounds.
Recent Publication: C. A. Bailey and B. L. Greene “A fluorometric assay for high-throughput phosphite quantitation in biological and environmental matricies” Analyst 2023, 148, 3650-3658.
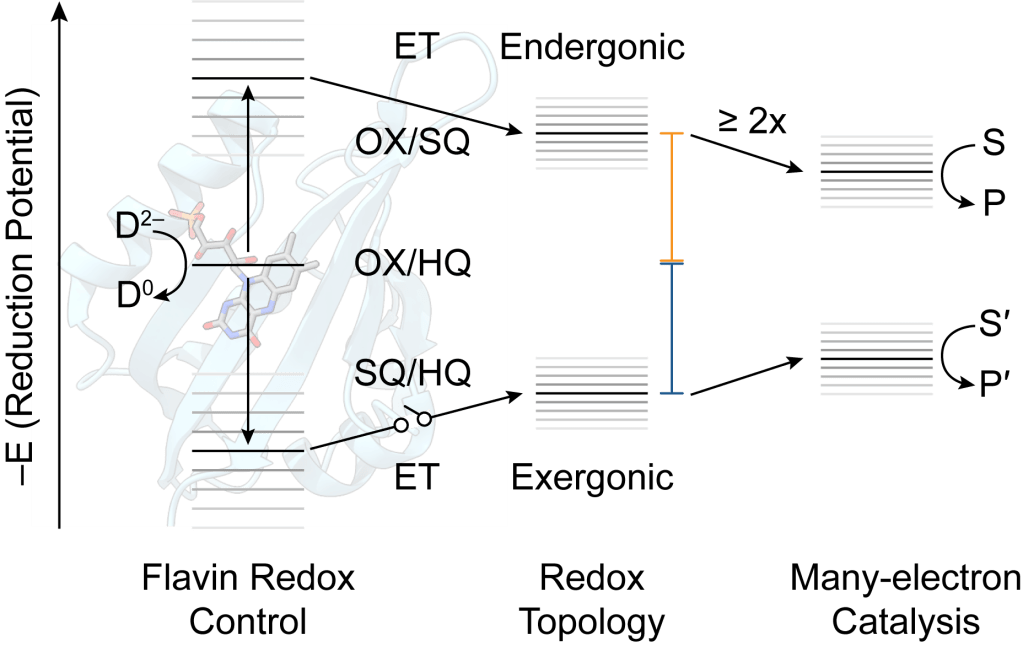
Electron bifurcation is a mechanism of biological energy conservation wherein exergonic (energy-releasing) electron transfer is conserved to drive pair-wise endergonic (energy-intensive) electron transfer. A molecular understanding of this phenomenon could enable metabolic engineering and increase electrocatalytic efficiency in biomimetic systems, yet the mechanism(s) of electron bifurcation remain opaque, particularly in the flavin-based electron bifurcases. We are using small flavoproteins, in a “bottom up” approach, to systematically evaluate how flavoproteins tune their cofactors for bifurcation, how redox topology affects bifurcation efficiency, and what are the pre-requisites for multi-turnover bifurcation and catalysis?
Recent Publication: B. L. Greene “Progress and opportunities in photochemical enzymology of oxidoreductases” ACS Catal. 2021, 11, 14635–14650.

Cysteine thiyl radicals are essential cofactors in a wide range of chemically challenging organic transformations, particularly in the anaerobic environment of the gut microbiome. Despite extensive study, these radicals are rarely observed directly and reactivity trends are challenging to construct, limiting our understanding of these important cofactors and our ability to target them therapeutically. We leverage (bio)chemical, spectroscopic, and computational tools to understand thiyl radical reactivity, with a particular focus on pyruvate formate lyase, an enzyme involved in anaerobic glycolysis in numerous opportunistic pathogens of the human gastrointestinal tract.
Recent Publication: J. C. Cáceres, A. Dolmatch, and B. L. Greene “The Mechanism of Inhibition of Pyruvate Formate Lyase by Methacrylate” J. Am. Chem. Soc. 2023, 145, 22504–22515.
